A full-stack institute for AI and biology research.
Headquartered in Palo Alto, California, Arc Institute is a nonprofit research organization founded on the belief that many important scientific programs can be enabled by new organizational models.
Arc’s mission is to accelerate scientific progress, understand the root causes of complex diseases, and narrow the gap between discoveries and impact on patients. Arc operates in partnership with Stanford University, UCSF, and UC Berkeley.
Arc provides scientists with multi-year funding to work on their most important ideas and invests in the rapid development of experimental and computational technological tools.
As individuals, Arc researchers collaborate across diverse disciplines to advance our understanding—and eventual treatment—of complex diseases, including cancer, neurodegeneration, and immune dysfunction.
Featured Research
See all research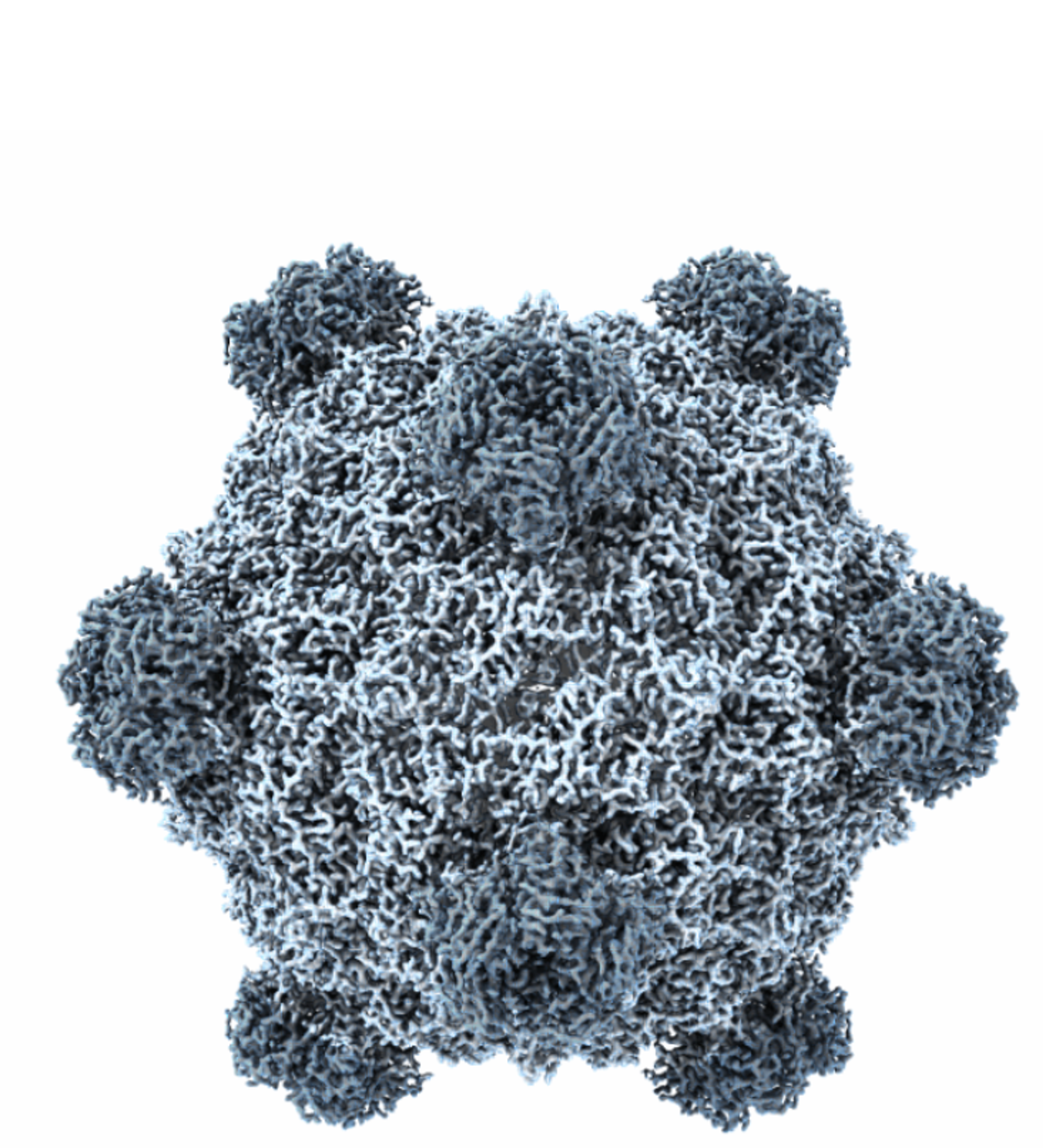
Generative design of bacteriophages with genome language models
Many important biological functions arise not from single genes but from complex interactions encoded by entire genomes. We report the first generative design of complete bacteriophage genomes using genome language models. We generated viable bacteriophages with target host tropism, using the phage ΦX174 as our design template.
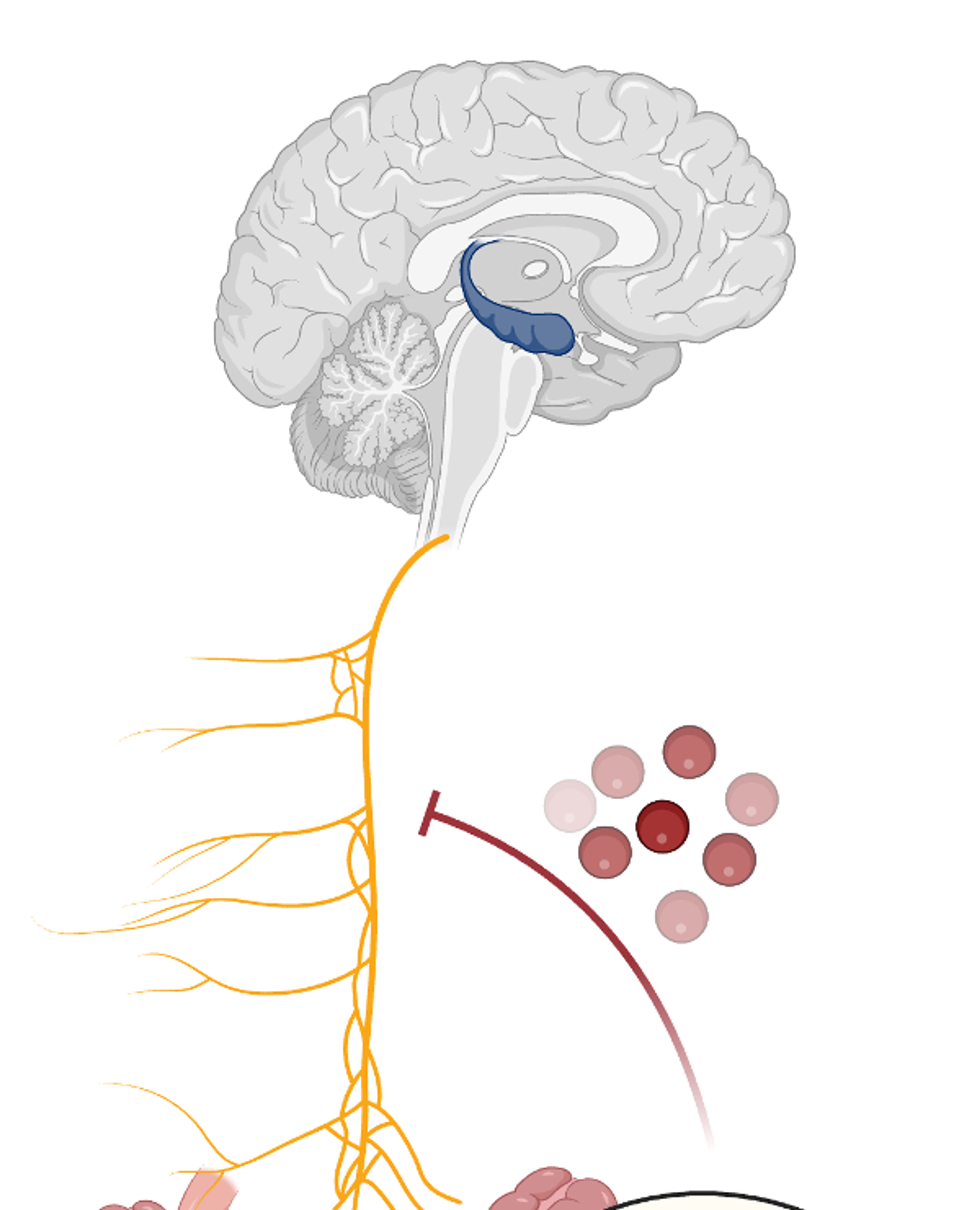
Intestinal interoceptive dysfunction drives age-associated cognitive decline
Here, by charting a high-resolution map of microbiome ageing and its functional consequences throughout the lifespan of mice, we identify a mechanism by which inhibition of gut–brain signalling during ageing results in impaired neuronal activation in the hippocampus and loss of memory encoding.
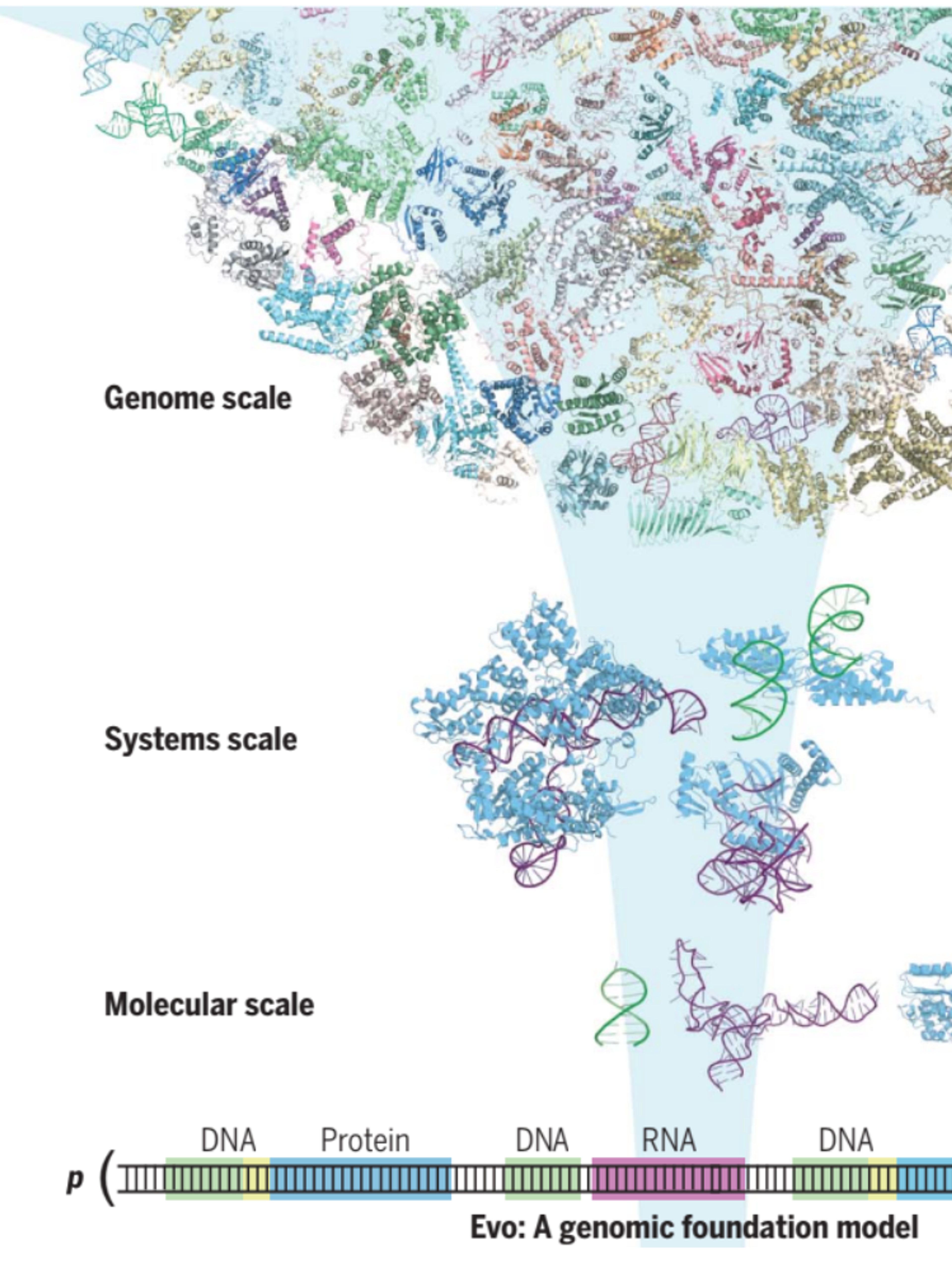
Genome modelling and design across all domains of life with Evo 2
AI models that learn information from genomic sequences have advanced prediction and design capabilities. Here we introduce Evo 2, a biological foundation model trained on 9 trillion DNA base pairs from a highly curated genomic atlas spanning all domains of life to have a 1 million token context window with single-nucleotide resolution.

Rapid directed evolution guided by protein language models and epistatic interactions
We present MULTI-evolve, a rapid evolution framework that systematically engineers multimutants. Our approach combines protein language models or existing functional data with epistatic modelling to predict synergistic combinations.

Vitamin B2 and B3 nutrigenomics reveals a therapy for NAXD disease
Vitamins are essential metabolites that must be obtained from external sources. In modern times, they have become widely available, leading to their ad hoc consumption. We developed a nutritional genomics framework to systematically identify monogenic diseases responsive to micronutrient modulation.
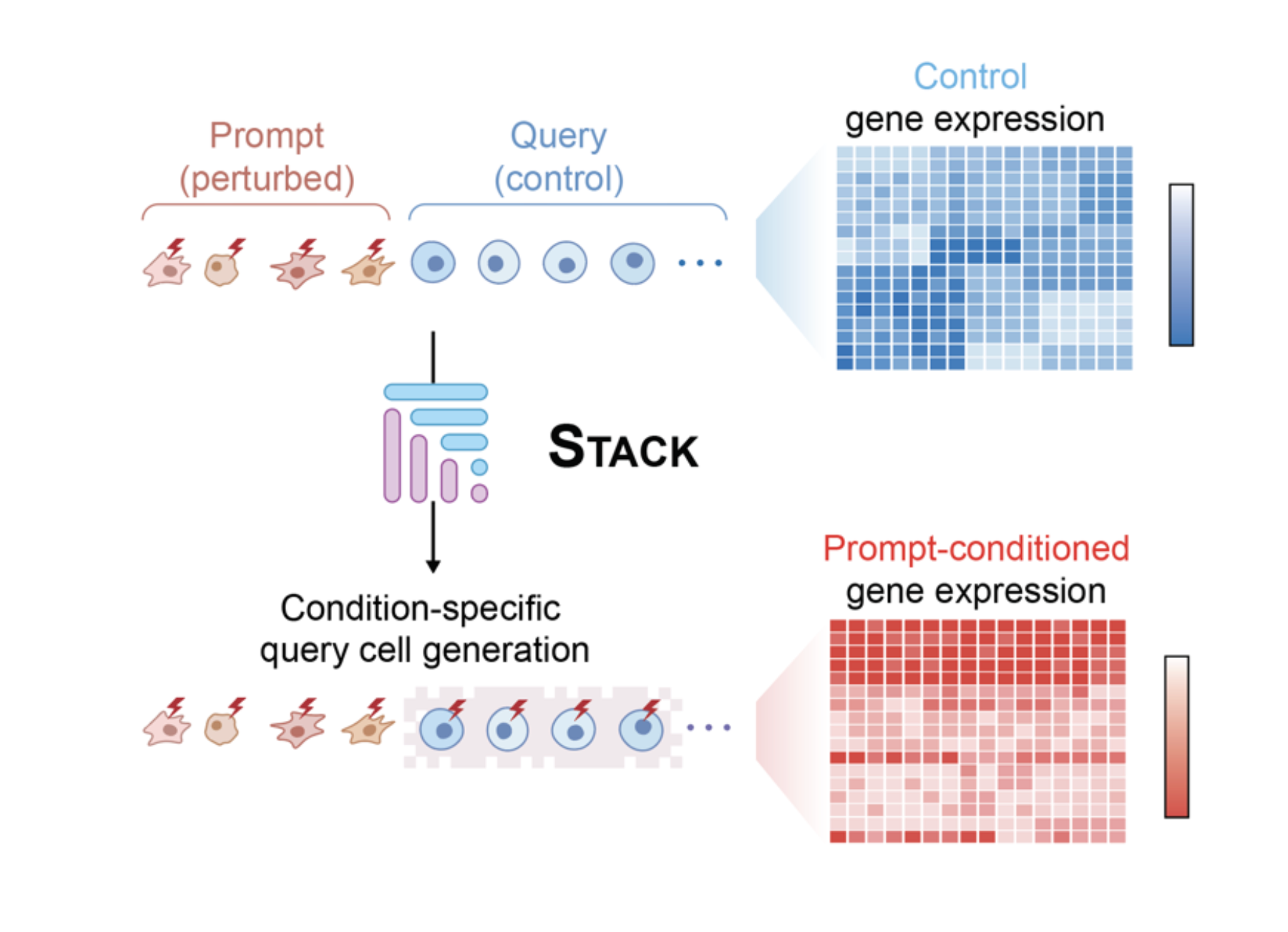
Stack: In-context learning of single-cell biology
Single-cell transcriptomics offers the promise of measuring the diversity of cellular phenotypes across species and diseases. Here, we present Stack, a foundation model trained on 149 million uniformly preprocessed human single cells that leverages tabular attention to generate representations for each cell informed by the cells in its context.
Arc's Model
More About Our Model

Join Arc Institute
Help us advance our mission to understand and treat complex human diseases.
Sign up to receive Arc Institute updates
Get Arc research delivered to your inbox:
Sign Up