
Patrick Hsu

Alison Fanton
The Therapeutic Potential of DNA Recombinases
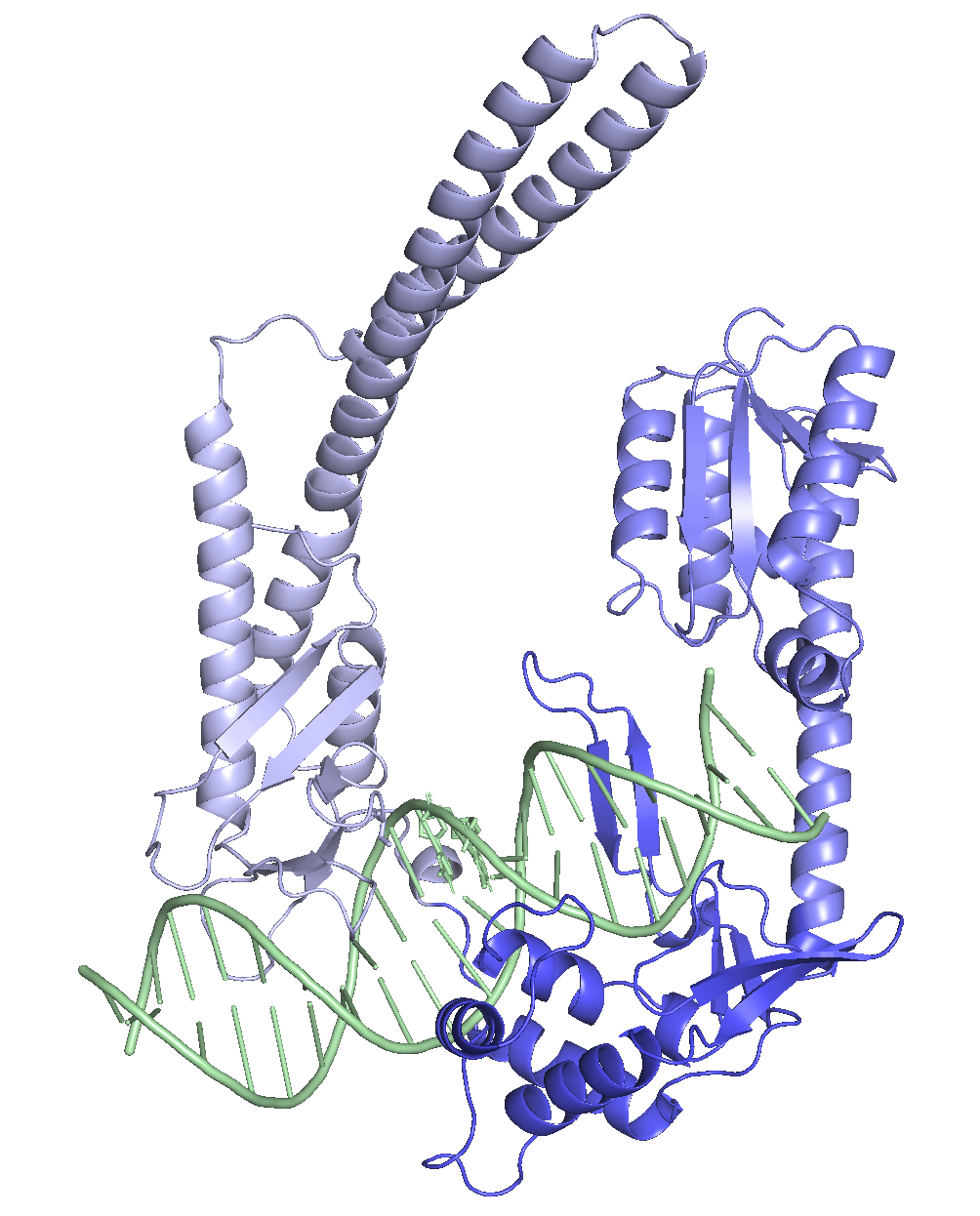
For decades, large serine recombinases (LSRs) have been known as the molecular machinery phages use to integrate their genomes into bacterial hosts. While systems like φC31 have been studied since the 1990s and were shown to work in human cells in 2000, LSRs never took off as genome editing tools the way CRISPR did. Naturally occurring LSRs have some serious trade-offs: some integrate into the human genome but target too many genomic sites, while others are highly specific but don’t target sequences that actually exist in human cells. Before we can use these as practical tools, both problems need to be simultaneously addressed.
Our lab's 2023 work, in collaboration with Ami Bhatt, Lacra Bintu, and Mike Bassik, screened over 60 different LSR orthologs to explore the diversity of their interactions with the human genome. We found that many systems recognized and integrated into endogenous human genome sequences with partial sequence similarity to their native attachment sites, termed pseudosites. However, specificity was a challenge. These enzymes were hitting tens to hundreds of pseudosites throughout the genome with varying efficiencies. So while we identified many new and improved LSR orthologs that recombine into the human genome, it was clear they needed serious engineering before becoming practical tools for precise genome editing.
In a paper published today in Nature Biotechnology titled "Site-specific DNA insertion into the human genome with engineered recombinases," we share the details and methodology of how we overcame these challenges.
Engineering Without Tradeoffs
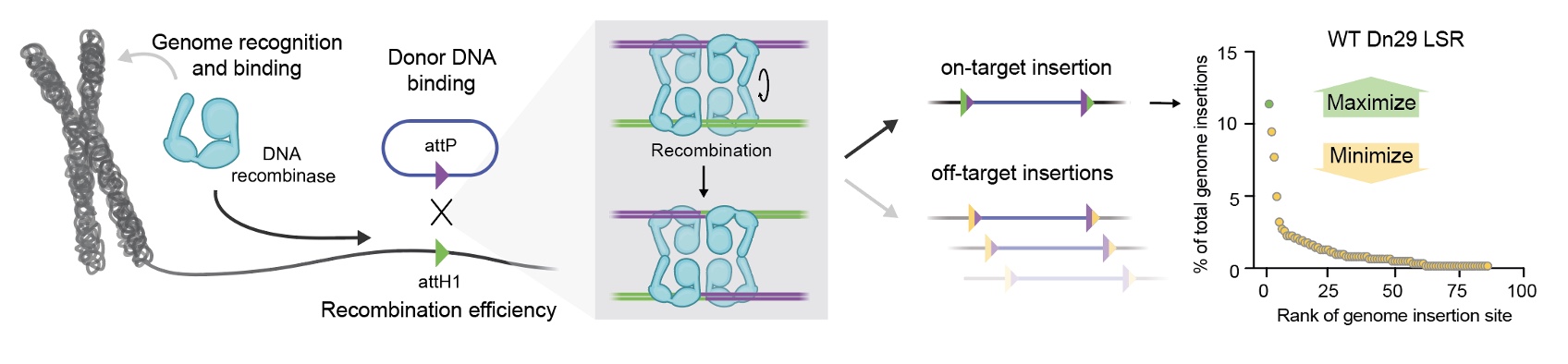
We started with Dn29, one of the LSR orthologs from our 2023 screen, because of its favorable integration characteristics. Dn29 has a 5% overall integration efficiency, and of those integrations, 12% go to a single site—we named attH1—located in an intron of NEBL. The rest are scattered across about 80 other off-target sites throughout the genome. But that middle-ground profile meant we had room to improve on both fronts.
Many engineering efforts that optimize only one metric simply trade one problem for another—gaining specificity at the cost of efficiency, or boosting activity while losing precision. We decided to optimize for two things at once: getting more insertions to attH1 and eliminating off-target sites.
To do this, we combined four distinct strategies that each tackled a different aspect of the problem:
-
Directed Evolution in Bacteria
We performed deep scanning mutagenesis of the entire Dn29 coding sequence and implemented a substrate-linked selection in E. coli that positively selected for active recombinants. After 12 evolution rounds with DNA shuffling to generate higher-order mutant combinations, we had a library rich in beneficial mutations. Crucially, we continuously validated promising bacterial variants in human cells, measuring both efficiency and specificity to ensure the resulting enzyme had therapeutic potential, not just higher activity in bacteria. -
Computational Modeling
Rather than exhaustively testing every possible mutant combination, we trained ridge regression models on single-mutant data to predict the activity of higher-order combinations, allowing us to prioritize variants for experimental validation. We were encouraged to find that LSR mutations are largely additive, enabling beneficial mutations to be combined. Structural modeling with AlphaFold3 revealed mechanistic insights into why these mutations were advantageous, showing mutational hot spots and residue charge patterns that could guide future engineering efforts. -
dCas9 Fusion
Fusing LSRs to catalytically dead Cas9 improved recombinase recruitment to target sites via sgRNAs, simultaneously enhancing efficiency and specificity. We pursued this strategy first because it was more tractable than launching a full directed evolution campaign, and it immediately paid off—our initial experiments showed 2–6-fold efficiency gains. We optimized sgRNA placement (~40 bp from the attachment site core), tested multiplexed guides, and incorporated sgRNA binding sites on donor plasmids for simultaneous genome and donor recruitment. -
Attachment Site Optimization
To develop the optimal plasmid substrate for Dn29, we performed deep mutational scanning of the attP sequence. This experiment highlighted the key nucleotides involved in protein–DNA interactions and identified optimized variants that enhanced integration efficiency.
After combining these independent engineering strategies into optimized constructs, we ended up with three key variants that each excel at different things:
- superDn29-dCas9: 53% integration efficiency (12-fold improvement over wildtype)
- hifiDn29-dCas9: 97% genome-wide specificity with 39% efficiency
- goldDn29-dCas9: Balanced profile at 90% specificity and 51% efficiency
One-Size-Fits-All Genome Editing
We tested the engineered recombinases across several different cell types to show they could handle diverse applications. In human embryonic stem cells, we hit 24% integration efficiency, and when we genotyped individual clones, 95% had precise heterozygous insertions at attH1. Primary T cells are notoriously harder to deliver double-stranded DNA templates, yet we still achieved 17% insertion efficiency using plasmid delivery, and 11% when we integrated a 5.8 kb CAR construct. We also tested non-dividing cells by treating them with a cell cycle-arresting compound, and still achieved 33% efficiency. The flexibility of these systems across such different cellular contexts demonstrates that LSRs can be robust genome engineering tools.
Beyond integrating DNA into the genome, we needed to know the cargo would retain functionality and was minimally disruptive to the function of other genes. We showed that attH1 integration maintains stable transgene expression through stem cell differentiation into hematopoietic progenitors. When we did bulk RNA-seq to look at off-target transcriptomic effects, the LSR-edited cells actually showed fewer differentially expressed genes than Cas9-edited cells at the same locus or at established safe harbors like AAVS1 and CLYBL. The integration is efficient, specific, and leaves the rest of the transcriptome relatively undisturbed.
Understanding potential safety concerns was also a priority. Across all variants, we saw no toxicity in cancer cells and low rates of indel formation. Compared to the natural Dn29 and Bxb1 (the current gold standard), the specificity enhanced variants also showed lower DNA damage response activation. While more extensive safety studies will be needed for therapeutic development, these initial results are very encouraging and support continued development.
Future Directions
LSRs recognize fixed DNA sequences, meaning they aren't reprogrammable like CRISPR or bridge recombinases (which our lab first described in 2024). However, for many therapeutic applications, this lack of programmability isn’t a limitation. The real advantages lie in the system’s simplicity, expanded cargo size, and robust and predictable edit outcomes. For genetic diseases with mutational heterogeneity across patient populations, using LSRs means you don't need to design a new therapeutic for each mutation. Instead, large cargo sizes enable replacing the entire gene with a corrective copy that includes proper regulatory elements. From a regulatory perspective, having a single therapeutic that works for all patients with a given disease, regardless of their specific mutation, is compelling.
Achieving this goal required developing a blueprint for engineering LSRs toward new genomic targets. The strategies we developed should be broadly applicable across the LSR family. For researchers wanting to try these tools, the system is remarkably simple. There are just two or three components: recombinase and donor DNA, and an added sgRNA for the optional fusion system. There's no dependence on endogenous repair machinery and no complex multi-component delivery or guide RNA design. We've tested them across multiple cell types with consistent success after establishing effective delivery protocols. If you're working with a cell line where transfection or electroporation already works, you should just try it.
The cross-species compatibility also matters for translation. We showed that Dn29 and goldDn29 recognize attH1-like sequences in marmosets, rhesus monkeys, cynomolgus monkeys, and mice, enabling straightforward preclinical studies.
Several areas still need development. Deep whole-genome sequencing will be necessary to detect ultra-rare rearrangements for clinical translation. Our framework can be extended to the thousands of mined, naturally occurring LSR orthologs to target desired sequences like AAVS1 or TRAC. And for sensitive cells like primary T cells, we need to optimize the trade-off between integration efficiency and cell survival, either through better donor delivery methods or continued protein engineering.
DNA recombinases offer a mechanistically distinct approach to genome engineering, allowing precise, large insertions without double-strand breaks. For research, these tools enable stable cell lines with near-clonal consistency in bulk populations. For therapeutics, they provide deliverable, precise integration of entire genes and regulatory elements. With continued development of characterization methods and exploration of natural recombinase diversity, we believe LSR-based genome engineering is moving from a promising approach to clinical reality.
Alison Fanton (X: @AlisonFanton) is a former graduate student in the Hsu lab. She received her PhD from the University of California, Berkeley in 2024.
Patrick Hsu (X: @pdhsu) is Co-Founder and a Core Investigator of Arc Institute and Assistant Professor of Bioengineering and Deb Faculty Fellow at the University of California, Berkeley.