


PerturbSpace brings spatial context to in vivo CRISPR screens
An additional labeling step transforms existing single-cell workflows into a spatial experiment

Cells don't behave in isolation. The identity of a cell, what it does, and how it responds to perturbation is shaped by where it sits in a tissue, what signals it receives from neighbors, and what its neighbors are doing. Single-cell RNA sequencing has given us a window into cellular identity and gene function, but the moment you dissociate a tissue, that spatial context is gone. You may learn what a cell is, but you lose where it is and what it was doing there.
Arc's In Vivo Functional Genomics team, part of the Mammalian Models Technology Center, runs CRISPR screens to generate high-quality tissue context data for our Virtual Cell Initiative. We need this data, and lots of it, to understand how gene knockouts change the structures cells build or how perturbations propagate to neighboring cells. But most existing spatial approaches require specialized instrumentation and complex workflows that aren't compatible with the scale of perturbation screening we need.
Multiple studies have sought to combine spatial profiling with CRISPR screens. Today we're sharing a preprint describing PerturbSpace, the approach we built to scale spatial CRISPR screens. PerturbSpace adds a single labeling step before a standard single-cell sequencing workflow. The output is a dataset where every cell carries its tissue location alongside its transcriptome, its CRISPR guide identity, and any other modalities your experiment includes. As a proof-of-concept, we processed 50 tissue sections spanning three whole spleens in a single day. An equivalent imaging-based approach would have taken months.
How PerturbSpace Works
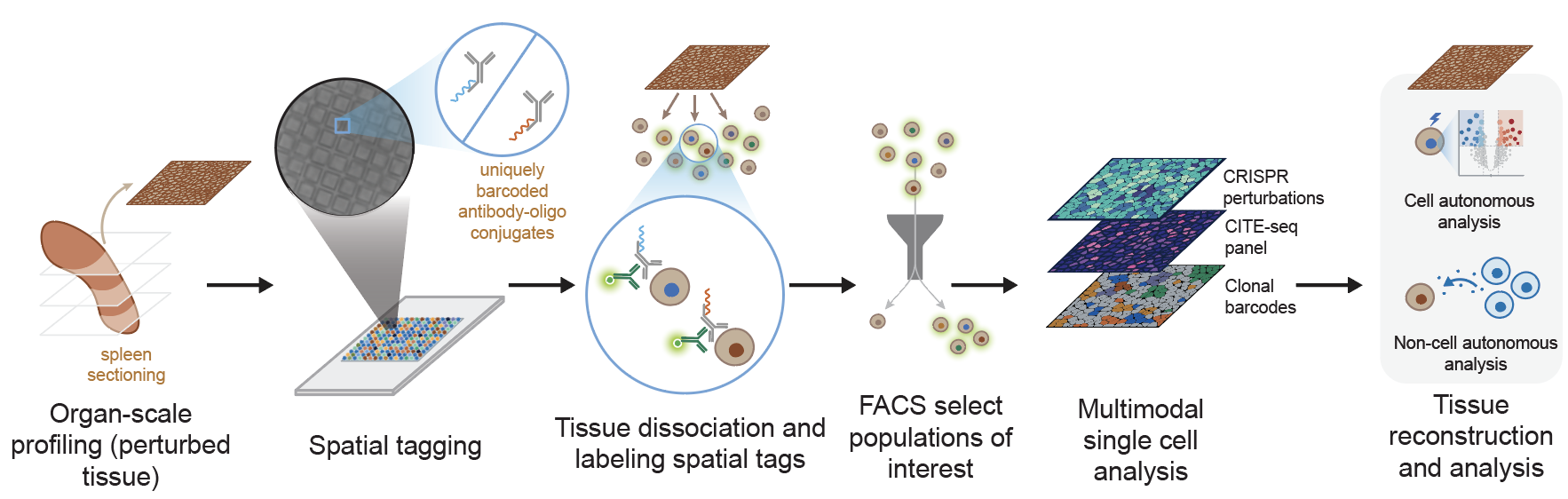
PerturbSpace achieves organ-scale spatial profiling in a single experiment using an antibody-based tissue hashing approach. The core of the method is a microwell array chip developed by Survey Genomics. Each well on the array is loaded with a unique spatial barcode and antibody-oligo conjugates that bind to the surface of any nucleated mammalian cell. To spatially profile a tissue, we section it, press it directly onto the chip for 30 minutes, and let the antibodies transfer their spatial barcodes onto whatever cells are sitting above each well.
We then lift the section off the array, dissociate it into a single-cell suspension, and sort the spatially labeled cells by FACS. This enrichment step, which is unique to PerturbSpace among spatial technologies, lets us select specific populations before sequencing. The sorted cells then go into a standard 10x Genomics single-cell sequencing workflow, with the spatial locations generated as a custom sequencing library.
Because PerturbSpace outputs a standard single-cell suspension, it's compatible with any readout that already works with single-cell sequencing (e.g., surface protein measurement via CITE-seq, chromatin accessibility via ATAC-seq, T and B cell receptor profiling, clonal barcoding). We demonstrated this by running spatial transcriptomics, a 119-protein CITE-seq panel, CRISPR perturbations, and clonal barcodes simultaneously from the same cells, with over 90% of cells mapped to a confident spatial neighborhood and sgRNA detection efficiency on par with standard Perturb-seq methods.
Mapping the Regenerating Spleen
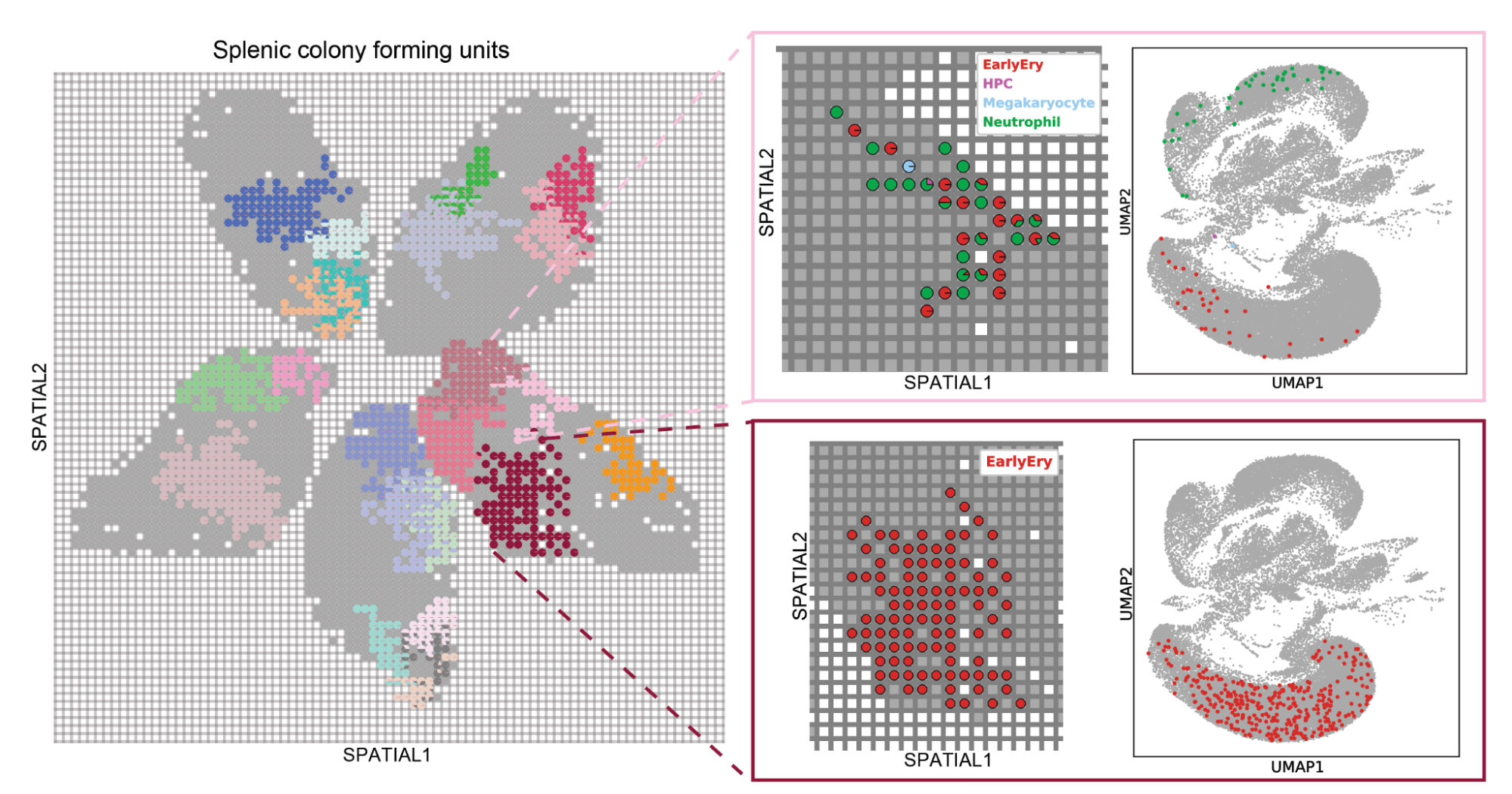
To test PerturbSpace, we applied it to regenerative hematopoiesis in the spleen. When hematopoietic progenitor cells engraft after transplant, each founding cell divides and differentiates in place, forming a splenic colony forming unit whose composition reflects the developmental potential of that founder. We labeled transplanted progenitors with CRISPR guides targeting 40 epigenetic regulators and unique clonal barcodes, so we could identify each colony in space and ask how perturbations changed their size, composition, and differentiation state. Across 50 spleen sections, we mapped 19,174 colonies.
We recovered known biology including Cebpa eliminating myeloid colonies entirely, and loss of Irf8 specifically ablated monocytic colonies. But what the spatial dimension of PerturbSpace revealed changes in colony size that is not possible with standard scRNA-seq. We were surprised to find that Rcor1 and Gltscr1 perturbations generated monocytic colonies significantly larger than controls, not because the cells were proliferating faster, but because both perturbations induced expression of cell-cell adhesion genes, apparently delaying the shedding of mature cells into the bloodstream. Hdac1 knockout produced smaller neutrophilic colonies driven by downregulation of the same adhesion programs. The spatial dimension pointed us toward a mechanism we wouldn't have thought to look for.
Perturbed Neighborhoods in the Liver
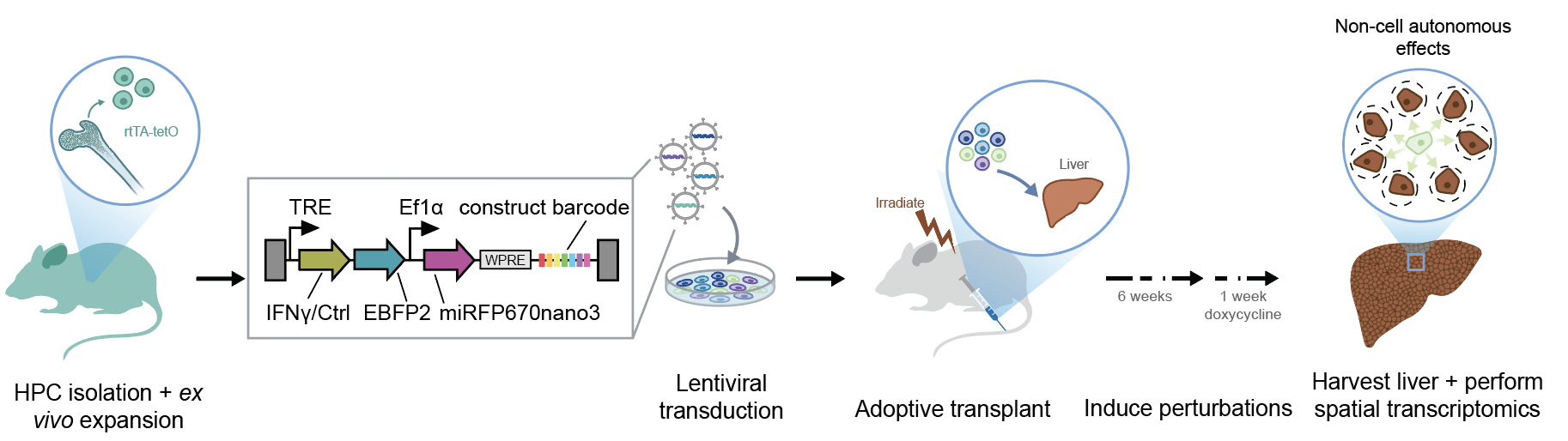
The spleen work asked how perturbations change the cells that carry them. We next looked in the liver to find out how perturbations in one cell type change the cells nearby.
We generated mice that express IFNγ or a control peptide in mature liver-resident immune cells, then used PerturbSpace to simultaneously profile both perturbed immune cells and all surrounding hepatic cell types. This way, we could directly compare the transcriptional state of bystander cells adjacent to perturbed immune cells against those in unperturbed neighborhoods.
Non-cell autonomous effects like these have been largely inaccessible to in vivo perturbation studies.
Try it Yourself
We are planning to continue our work to apply PerturbSpace, and hope other labs can take advantage of it too. A few things worth sharing before you get started.
Because it requires dissociation into live single-cell suspensions, PerturbSpace isn't immediately compatible with some cell types that don't tolerate standard single-cell RNA-seq. The spatial resolution is also neighborhood-level rather than single-cell, which means cellular morphology and precise positioning are outside what PerturbSpace can capture. But for most questions about tissue organization, gene function, and cell-cell communication, we feel that tradeoff is worth it.
Learn more about PerturbSpace in the recording below. Please reach out if you have any questions or are interested in collaborating.
###
Nevue, A.A., Hartoularos, G.C., De Valle, C., Ramachandran, K., Barron, J.J., Lee, H., Calleja Cervantes, M.E., Bowness, J., Velten, L., Ricci-Tam, C., Dobin, A., Levy, M., Averbukh, I., Lara-Astiaso, D. (2026). Spatially resolved, multimodal in vivo Perturb-seq using antibody-based cell hashing. BioRxiv. https://doi.org/10.64898/2026.05.25.727765
Alexander Nevue is a Senior Scientist on the In Vivo Functional Genomics team at Arc Institute.
Inna Averbukh is a Senior Scientist on the Bioinformatics team at Arc Institute.
David Lara-Astiaso leads the In Vivo Functional Genomics group within Arc's Mammalian Models Technology Center.